### Life 708
***
# Genome Assembly
***
### Will Rowe
View the presentation online at:
[will-rowe.github.io/genome-assembly-2017](https://will-rowe.github.io/genome-assembly-2017)
---
## Aims of these slides
***
* introduction to genome assembly
* understand how some assembly algorithms work
* how to assess assembly quality
* walkthrough a typical genome assembly workflow
----
## Introduction
***
* no technology yet exists to sequence **full-length** DNA
* we have to sequence short fragments and piece them together computationally
* assembly is the process of reconstructing a genome sequence from raw sequencings reads
* there are two types of genome assembly
- *de novo*
- re-sequencing (reference-guided)
* we'll focus on de novo assembly of bacterial genomes
---
### Why assemble genomes?
***
* we need a reference genome (we might not already have one)
* we want to look at genome structure / put features into context
* to make comparisons to other genomes
---
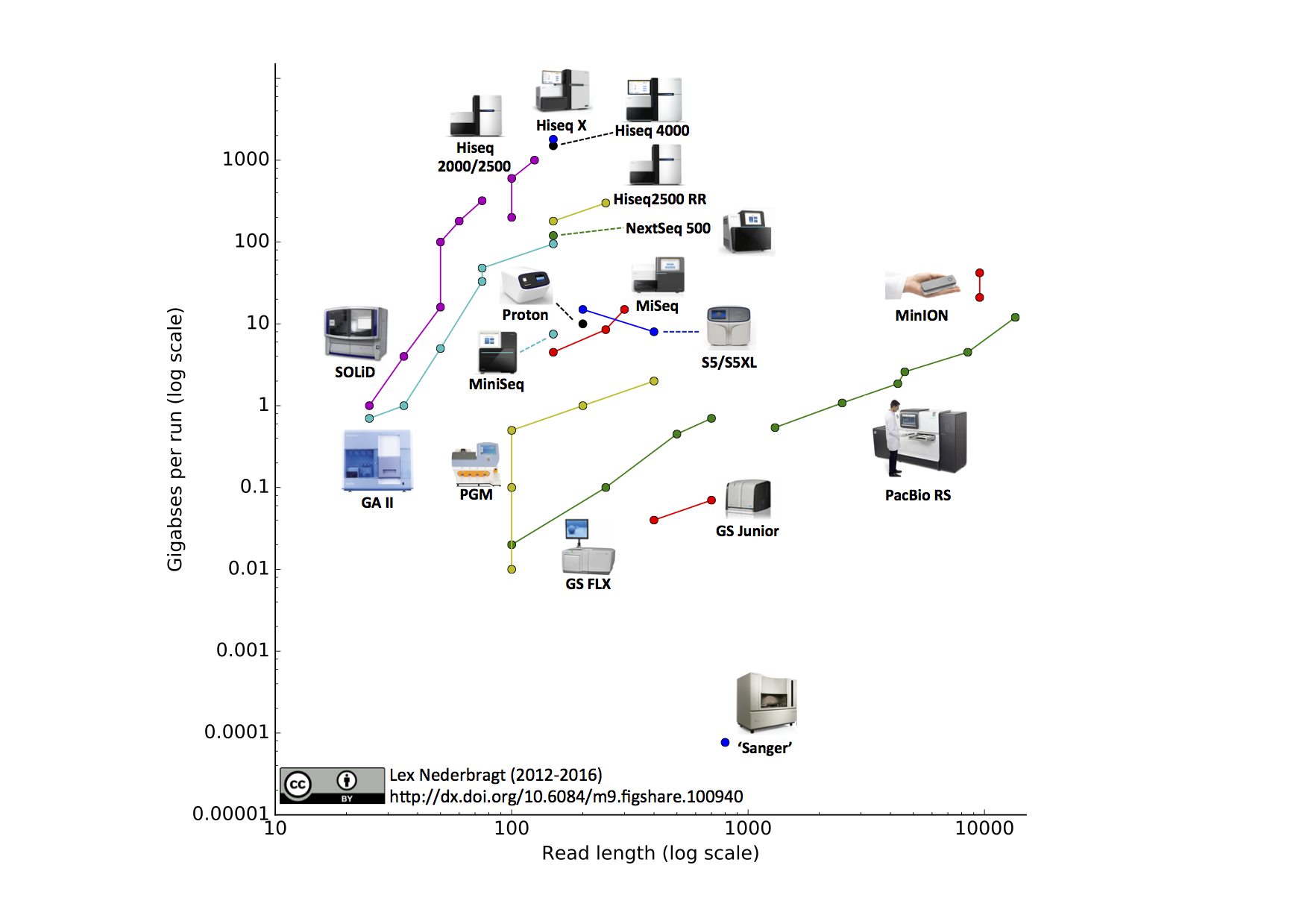
* there are many different sequencing platforms available
- you've covered this in LIFE708 already
- they each have advantages / disadvantages
* platform choice is dictated by experimental design
- there are also cost/practical considerations
* there are different assembly approaches
- e.g. for long / short read platforms
* sequencing bias and sequencing error are factors that may impact our assembly quality
---
---
### *De novo* assembly
***
* the DNA read by sequencers has been randomly fragmented and amplified as part of library preparation\*
* sequencing reads are therefore unordered fragments of a genome
* the aim of *de novo* assembly is to piece the reads together so that they give the whole genome sequence
* assembly quality is dictated by several factors
- read length and **coverage**
- sequence data quality
- genome complexity
---
* sequencing **coverage** is the average number of reads that align to known reference bases
* low sequencing coverage can result in genomic regions with no sequencing reads
* appropriate read length depends on genome size - i.e. reads need to be long enough to contain unique features
- genomic repeats make assembly hard
- hard to resolve if reads are shorter than repeat length
* paired-end reads with a long insert size can address read length limitations (i.e. resolve repeats)
---
### Let's make sure we understand some terminology
***
|term|definition|
|-------|-------|
|assembly|reconstructing a genome sequence from raw sequencings reads|
|read|fragments of our genome generated by a sequencer|
|coverage|the average number of reads that align to known reference bases|
|contig|a *contigious sequence* built from overlapping reads and representing a consensus region of DNA|
|scaffold|sets of non-overlapping contigs separated by gaps of known length|
|algorithm|a set of rules to perform a task|
|graph|represents relationships using nodes and edges|
---

----
## Aims of these slides
***
* introduction to genome assembly ✓
* understand how some assembly algorithms work
* how to assess assembly quality
* walkthrough a typical genome assembly workflow
----
## Assembly algorithms
***
* it's important to know how genome assemblers work
- very important for experimental design
- helps us to choose the right tool for assembling data
- allows us to interpret results properly!
- we can also troubleshoot if our assembly isn't as expected
---
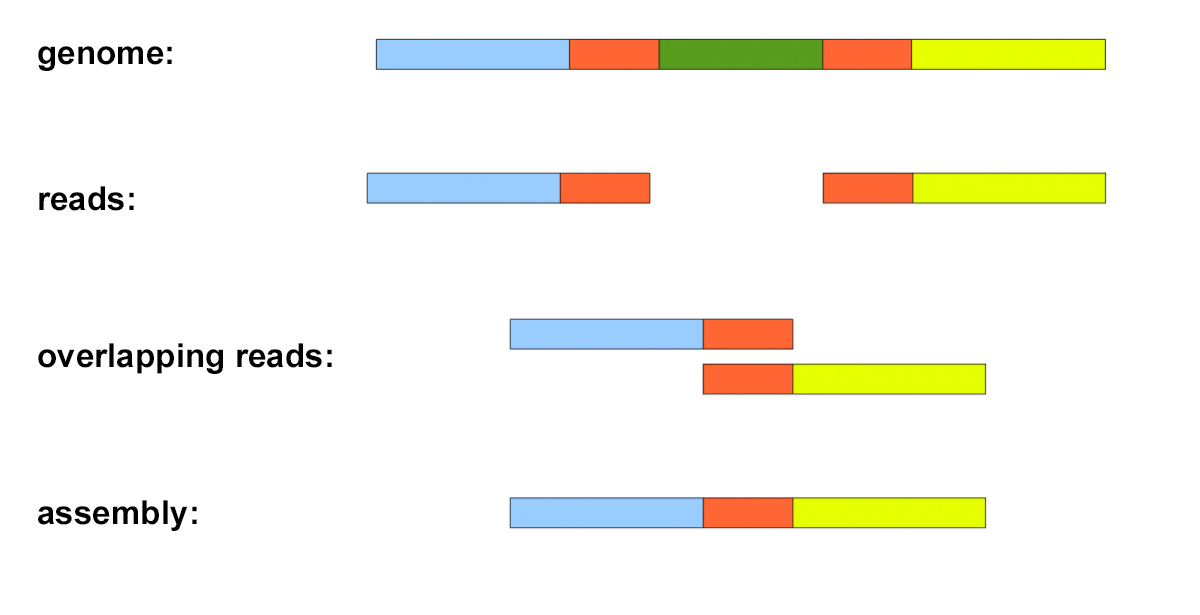
### Greedy algorithm
***
* performs pairwise comparison of reads
* combines any read pairs with sufficient overlap between edges
* assembly stops when no more overlaps are found
* greedy algorithms aren't good with repeats - leading to misassemblies
* they also can't easily utilise global information (e.g. read pairs)
---
### Greedy algorithm
***
---
### Graph-based assembly
***
* represent relationships between sequenced reads
* graphs consist of edges and nodes
- graph can be directed or undirected (edge directionality)
- nodes can have multiple edges
- assemblers generally use directed multigraphs
* most modern genome assemblers use these graphs
- build graph from reads and traverse it to derive genome
* several graph-based assembly algorithms
- **overlap layout consensus (OLC)** - reads remain intact
- **de Bruijn graph** - breaks down reads to k-mers and finds exact overlaps
- **string graph** - variant of OLC approach
- **hybrid** - combines multiple approaches / sequence data
---
### Graph theory: the Bridges of Konigsberg
***

* can we visit each part of the city by crossing each bridge once?
---
### Graph theory: the Bridges of Konigsberg
***
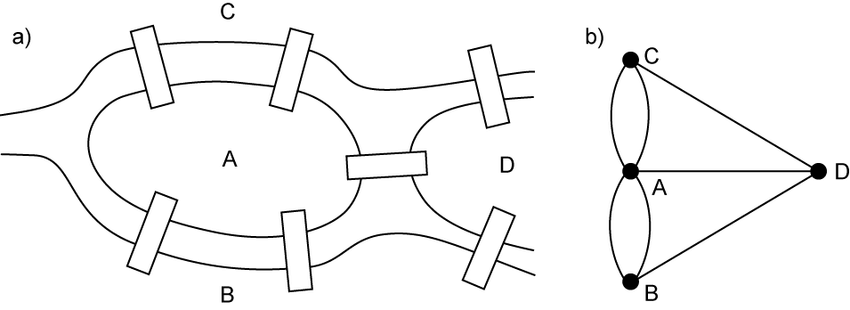
* represent each land mass as a node and each bridge as an edge
* Eulerian path = visit every edge of a graph exactly once
* it's not possible in this case!
---
### Graph theory: Eulerian paths
***
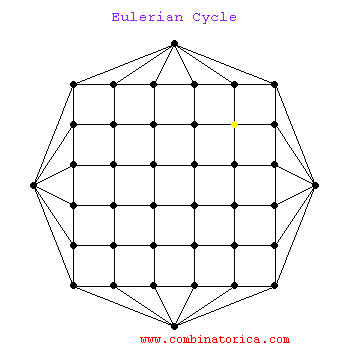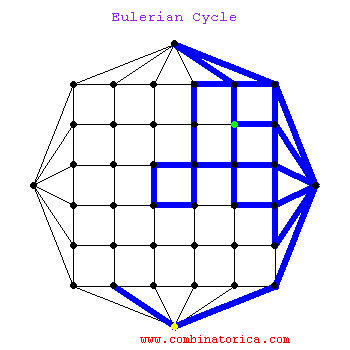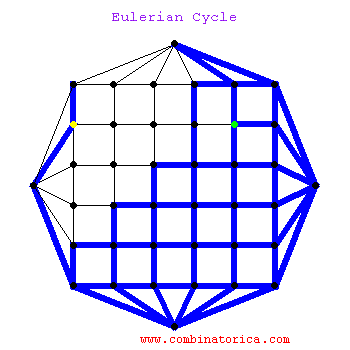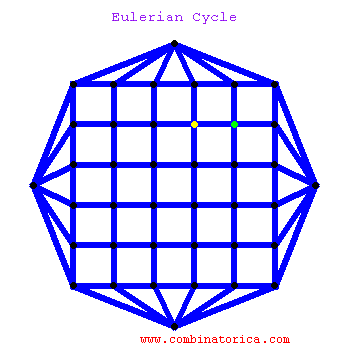
* graphs with Eulerian paths are termed Eulerian
* traversing a Eulerian path takes linear time
---
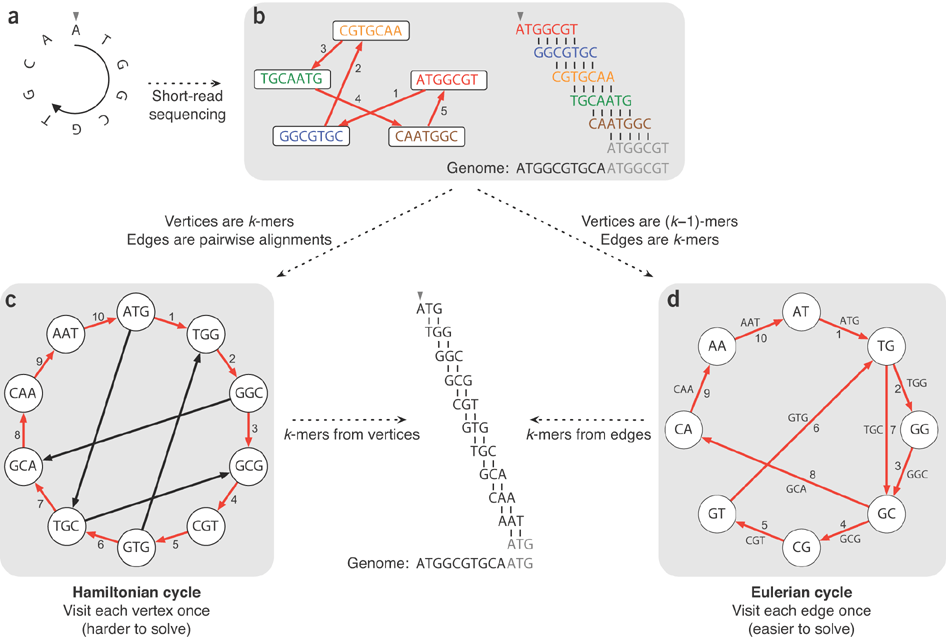
### Graph theory: Hamiltonian paths
***
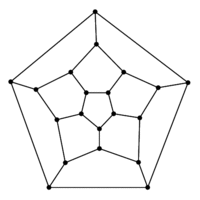
* Hamiltonian path = visit every node of a graph exactly once
* traversing a Hamiltonian path is hard to implement an algorithm for (NP-complete)
---
### Graph theory: Hamiltonian paths
***
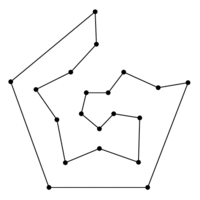
* visit every node once
* traversing a Hamiltonian path is hard to implement an algorithm for (NP-complete)
---
### Overlap-Layout-Consensus (OLC) algorithm
***
* based on the greedy algorithm but finds all possible overlaps
* finds **overlaps** (aligning the reads) & constructs overlap graph
* **layout** reads according to alignment (find Hamiltonian path)
* gets the **consensus** by traversing Hamiltonian path
* E.G. assemblers: Celera, Newbler, Canu
---
### Overlap-Layout-Consensus (OLC) algorithm
***
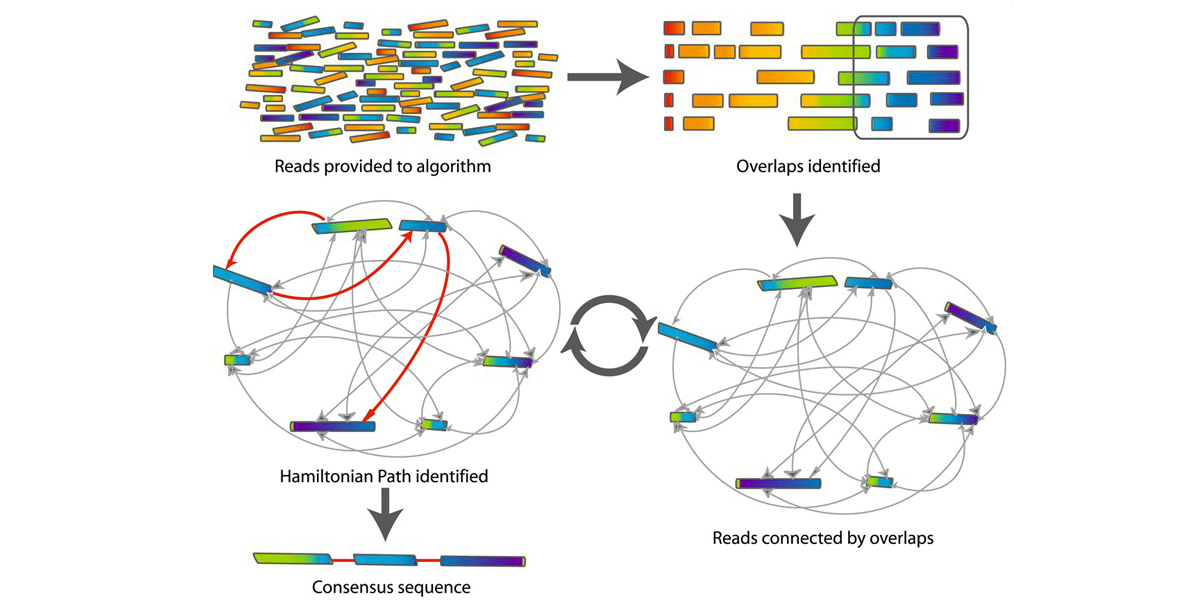
---
### De Bruijn graph-based algorithm
***
* reads are broken down to k-mers (substrings of length k)
- why do we use k-mers and not reads?
* a de Bruijn graph is constructed from the k-mers
- k-mers are connected if they have k-1 shared bases
* the genome is derived using the Eulerian path through the graph
* assuming no sequencing error, sequencing reads will match the genome perfectly
- therefore the de Bruijn graph constructed from reads will be the same as that constructed from the underlying genome
* E.G. assemblers: SPAdes, Velvet, ABySS
---
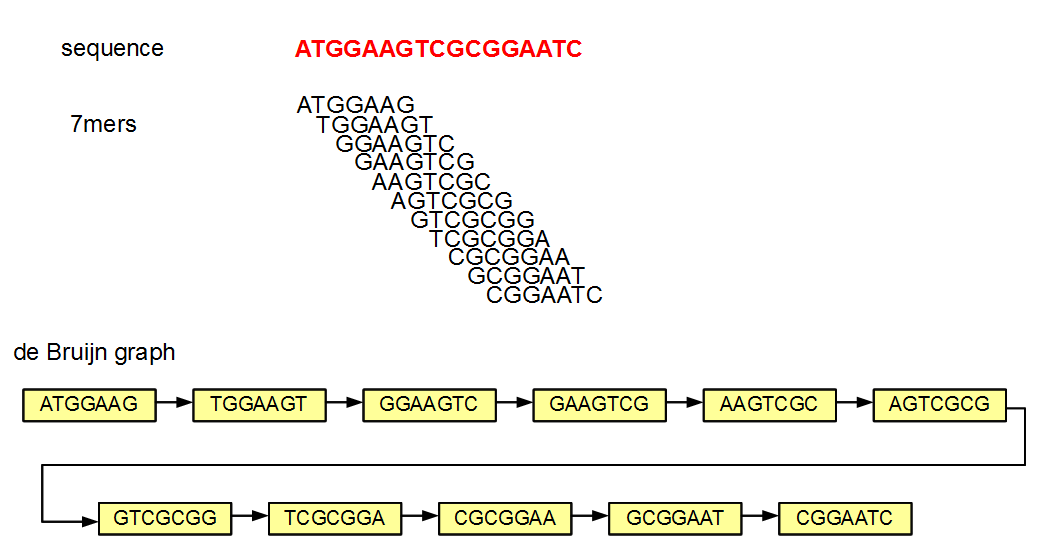
* edges connect nodes that have overlaps of 6 (=k-1) bases
* in this example, no k-mers appear multiple times
---
---
### De Bruijn graph-based algorithm: considerations
***
* we have seen that repetitive regions lead to multiple edges
* DNA strandedness will also increase number of node edges (think reverse complementing)
* how are sequencing errors and artifacts handled?
* what k-mer size should we use?
---
### De Bruijn graph-based algorithm: graph features
***
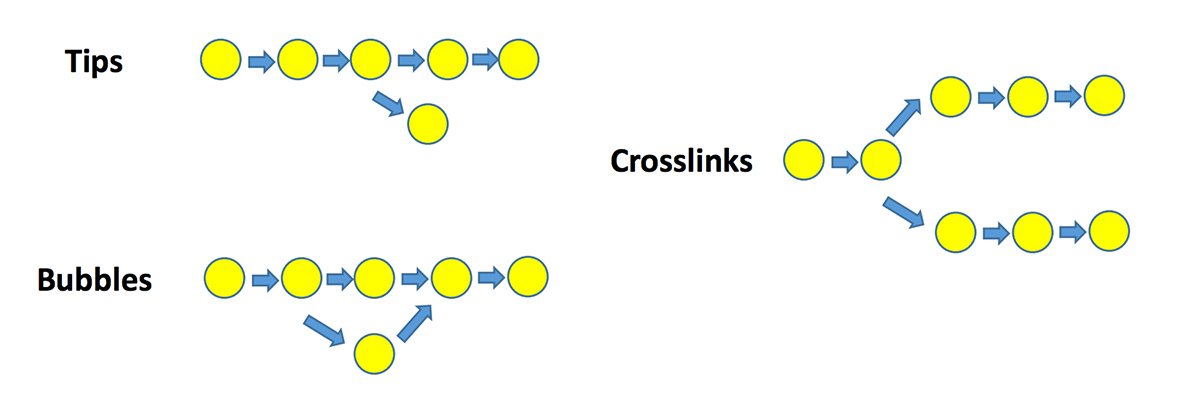
* use k-mer frequency to resolve these graph features
- remove low depth kmers
- clip tips, merge bubbles, remove links
- resolve small repeats using long k-mers
---
### De Bruijn graph-based algorithm: k-mer size
***
* avoid using an even numbered k-mer size
- they can lead to reverse complementing
- affects the strand specificity of the graph
- palindromic k-mers are avoided with an odd k
* increasing k-mer size can resolve ambiguities
- higher k-mer size can < number of edges and < possible paths
- however, higher k-mer size also more sensitive to sequencing errors
- higher k-mer size means more RAM needed
* try several k-mer sizes to get the best assembly!
---
### De Bruijn graph-based algorithm: recommended resources
***
good websites and papers:
[coding4medicine](https://www.coding4medicine.com/Members/Materials/assembly/files/chapter1.html), [Langmead lab teaching materials](http://www.langmead-lab.org/teaching-materials/), [Compeau et al. 2011](http://www.nature.com/nbt/journal/v29/n11/full/nbt.2023.html), [EBI course](https://www.ebi.ac.uk/training/online/course/ebi-next-generation-sequencing-practical-course/part-1)
great explanation video:
---
### Recap:
---
## Practical assembly example
***
Theory is one thing but let's look at implementing an assembly algorithm ourselves
Example found here: [bits of bioinformatics blog](https://pmelsted.wordpress.com/2013/11/23/naive-python-implementation-of-a-de-bruijn-graph/)
---
----
## Aims of these slides
***
* introduction to genome assembly ✓
* understand how some assembly algorithms work ✓
* how to assess assembly quality
* walkthrough a typical genome assembly workflow
----
## Assembly quality
***
We assess quality by looking at assembly **contiguity**, **completeness** and **correctness**
---
### Contiguity
***
* Ideally, we want very long contigs and few of them
* We measure contiguity using:
- contig number
- contig length (average, median and maximum)
- N statistics (e.g. N50)
* N50 is a statistical measure of the average length of a set of contigs
- 50% of the entire assembly is contained in contigs => to the N50 value
---
### Completeness and correctness
***
* completeness is the proportion of the sequenced genome represented by the assembled contigs
- completeness = assembled genome size / estimated genome size
* correctness is a measure of the number of errors in the assembly
- feature compressions (i.e. repeats)
- improper contig scaffolding
- introduced SNPs/InDels
---
### Assessing assembly quality: checklist
***
* check the three C's - contiguity, completeness, correctness
* look at all the statistics we are given by assemblers
* map the original reads to the assemblies to find inconsistencies
* check / visualise against other assemblies / references
----
## Aims of these slides
***
* introduction to genome assembly ✓
* understand how some assembly algorithms work ✓
* how to assess assembly quality ✓
* walkthrough a typical genome assembly workflow
----
## Typical genome assembly workflow
***
* experimental design
- genome size? repetitive regions? plasmids?
* DNA extraction
- how much DNA? quality?
* library preparation & sequencing
- library design (paired-end/mate-pair, insert size)? how much data needed?
* quality assessment of sequence libraries
- how to we assess quality before/after sequencing?
* genome assembly
- how to validate / compare / improve / annotate?
---
### DNA extraction
***
* we want high molecular weight DNA with no contamination
* use absorption ratios to estimate quality and quantity (Qubit, Nanodrop etc.)
* run the samples on a gel to check for degradation, protein / RNA contamination etc.
---

---
### Library preparation and sequencing
***
You've already covered technologies - which one to choose?
How many reads do I need to get a good assembly?
---
### Library preparation and sequencing
***
* for bacterial genome assembly, we usually want at least 30X sequencing coverage
* **sequencing coverage** is the average number of reads that align to known reference bases
* the Lander/Waterman equation is a method to compute expected coverage
## coverage = (no. reads * length of read) / genome size
---
### Example calculation
***
* an Illumina MiSeq flowcell can generate 25 million reads
* organism genome size: 4.5 Mbp (Megabase pair)
* required coverage: x30
* read length: 150 bp
* number of reads needed (N):
N = (30 x 4.5e+6) / 150
N = 9e+5 reads
---
### Quality assessment
***
* once we have sequence data, we want to check the quality
- we'll be covering how to check sequence read quality tomorrow
* example FASTQC report [found here](https://willrowe.net/_slides/slide-data/genome-assembly/QC/multiqc_report.html)
---
### Assemble!
***
In the practical tomorrow we will use a real-world *Shigella flexneri* sequencing dataset
We will be retrieving the data, quality checking it and then running some assembly algorithms. We'll then assess our assemblies, compare them to a reference genome and annotate our draft assemblies.
---
### Assembly annotation
***
* once we have assembled, we want to extract information from the genome
* covering annotation would be a lecture in itself
* the basic levels are: nucleotide, protein and process
- identify known features using alignment
- prediction of new features
* annotation is an important part of comparative genomics and was covered in LIFE721
---
### Closing the genome
***
* we have described how to assemble a **draft** genome
* draft assemblies contain errors
- sequence errors
- misassemblies
- contamination
* manual curation, re-sequencing and other approaches are often needed to close a genome
----
## Aims of these slides
***
* introduction to genome assembly ✓
* understand how some assembly algorithms work ✓
* how to assess assembly quality ✓
* walkthrough a typical genome assembly workflow ✓
----
## References used in this slide deck
***
https://flxlexblog.files.wordpress.com/
http://math.nmsu.edu/
https://link.springer.com/article/10.1007/s12575-009-9004-1
http://www.homolog.us/Tutorials
http://cbsuss05.tc.cornell.edu/doc/
http://debruijn.herokuapp.com/
https://pmelsted.wordpress.com/
https://www.slideshare.net/torstenseemann/de-novo-genome-assembly-tseemann-imb-winter-school-2016-brisbane-au-4-july-2016
https://genomics.ed.ac.uk/